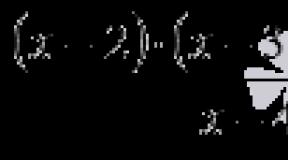Алкены II.Окислительное расщепление алкенов. Материал для подготовки к егэ (гиа) по химии (10 класс) на тему: расстановка коэффициентов в уравнениях реакций окисления алкенов перманганатом калия, протекающих в кислой среде с разрывом углеродной цепи Окисл
4.5.б. Окислительное расщепление алкенов
При окислении алкенов щелочным водным раствором перманганата калия при нагревании или раствором KMnO 4 в водной серной кислоте, а также при окислении алкенов раствором оксида хрома (VI) CrO 3 в уксусной кислоте или дихроматом калия и серной кислотой первоначально образующийся гликоль подвергается окислительной деструкции. Конечным результатом является расщепление углеродного скелета по месту двойной связи и образование в качестве конечных продуктов кетонов и (или) карбоновых кислот в зависимости от заместителей при двойной связи. Если оба атома углерода при двойной связи содержат только по одной алкильной группе, конечным продуктом исчерпывающего окисления будет смесь карбоновых кислот, тетразамещенный при двойной связи алкен окисляется до двух кетонов. Однозамещанные алкены с концевой двойной связью расщепляются до карбоновой кислоты и углекислого газа.

Из-за невысоких выходов карбоновых кислот и кетонов, реакции исчерпывающего окисления алкенов в классическом варианте не нашли широкого применения и ранее использовались, в основном, для установления строения исходного алкена по продуктам деструктивного окисления. В настоящее время окисление алкенов (R-CH=CH-R и R-CH=CH 2) до карбоновых кислот (RCOOH) с помощью перманганата или дихромата калия проводят в условиях межфазного катализа. Выходы карбоновых кислот при этом превышают 90%.

4.5.в. Озонолиз алкенов
Реакция алкенов с озоном является наиболее важным методом окислительного расщепления алкенов по двойной связи. В течение многих десятилетий эта реакция служила основным методом определения строения исходного углеводорода, а также находила применение в синтезе разнообразных карбонильных соединений. Реакция алкена с озоном проводится пропусканием тока ~5%-ной смеси озона и кислорода в раствор алкена в хлористом метилене или этилацетате при -80 0 -100 0 С. Окончание реакции контролируется пробой на свободный озон с иодидом калия. Механизм этой своеобразной и сложной реакции установлен главным образом благодаря работам Р Криге. Первым продуктом 1,3-диполярного циклоприсоединения к двойной связи является так называемый мольозонид (1,2,3-триоксолан). Этот аддукт нестабилен и далее самопроизвольно разлагается с раскрытием цикла и образованием в качестве конечного продукта нормального озонида (1,2,4-триоксолана).

В настоящее время общепризнано, что превращение мольозонида в обычный озонид происходит по механизму расщепления - рекомбинации. Мольозонид претерпевает самопроизвольное раскрытие нестабильного 1,2,3-триоксоланового цикла с образованием карбонильного соединения и биполярного иона, которые далее реагируют между собой также по схеме 1,3-диполярного циклоприсоединения.

Приведенная схема перегруппировки мольозонида в нормальный озонид подтверждается тем, что если до полного образования озонида в реакционной смеси присутствует в качестве "перехватчика" биполярного иона другое карбонильное соединение, то образуется так называемый "смешанный озонид". Так, например, при озонилизе цис -стильбена в присутствии бензальдегида, меченного изотопом 18 О, метка входит в состав эфирного, а не перекисного мостика озонида:
Этот результат хорошо согласуется с образованием смешанного озонида при рекомбинации биполярного иона с меченным бензальдегидом:

Озониды представляют собой очень нестабильные соединения, разлагающиеся со взрывом. Их не выделяют в индивидуальном виде, а расщепляют при действии самых разнообразных регентов. Следует различать восстановительное и окислительное расщепление. При гидролизе озониды медленно расщепляются на карбонильные соединения и перекись водорода. Перекись водорода окисляет альдегиды до карбоновых кислот. Это так называемое окислительное разложение озонидов:

Таким образом, при окислительном разложении озонидов образуются карбоновые кислоты и (или) кетоны в зависимости от строения исходного алкена. В качестве окислителей можно использовать кислород воздуха, перекись водорода, перкислоты или гидроокись серебра. Наиболее часто в синтетической практике для этой цели используют перекись водорода в уксусной или муравьиной кислоте, а также перекись водорода в щелочной среде.


На практике метод окислительного разложения озонидов используется, в основном, для получения карбоновых кислот.
Более важное значение имеет восстановительное расщепление озонидов. В качестве восстановителей наиболее часто используются цинк и уксусная кислота, трифенилфосфин или диметилсульфид. В этом случае конечными продуктами озонолиза оказываются альдегиды или кетоны в зависимости от строения исходного алкена.



Из приведенных выше примеров видно, что тетразамещенный при двойной связи алкен при озонолизе и последующем восстановительном разложении озонида образует два кетона, тогда как тризамещенный алкен дает кетон и альдегид. Дизамещенный симметричный алкен при озонолизе образует два альдегида, а алкены с концевой связью - альдегид и формальдегид.
Интересной модификацией озонолиза является метод, где в качестве восстановителя озонида используется боргидрид натрия, В этом случае конечными продуктами реакции оказываются первичные или вторичные спирты, образующиеся при восстановлении соответственно альдегидов и кстонов.


Озонолиз алкенов - это сложный, трудоемкий и взрывоопасный процесс, требующий применения специальной аппаратуры. По этой причине были разработаны другие методы окислительного расщепления алкенов до карбонильных соединений и карбоновых кислот, которые с успехом заменяют реакцию озонолиза в синтетической практике.
Один из современных препаративных методов окислительной деструкции алкенов был предложен в 1955 г Р. Лемье. В основе этого метода лежит гидроксилирование алкенов с помощью перманганата калия с последующим расщеплением вицинального гликоля периодатом натрия NaIO 4 при рН ~ 7 8. Периодат сам по себе не взаимодействует с алкеном. Продуктами этого двухстадийного окислительного расщепления являются кетоны или карбоновые кислоты, поскольку альдегиды в этих условиях также окисляются до карбоновых кислот. В методе Лемье не возникает трудоемкой проблемы отделения одного из продуктов реакции, - двуокиси марганца, так как и двуокись, и манганат вновь окисляются периодатом до перманганат-иона. Это позволяет использовать только каталитические количества перманганата калия. Ниже приведены некоторые типичные примеры окислительного расщепления алкенов по методу Лемье.

Цитронеллол - спирт, входящий в состав розового масла, масла герани и лимона, - окисляется смесью перманганата калия и периодата натрия в водном ацетоне при 5 10 0 С до 6-гидрокси-4-метилгексанкарбоновой кислоты с количественным выходом.

В другой разновидности этого метода вместо перманганата калия используют каталитические количества тетраоксида осмия (Лемье, Джонсон 1956 г). Особое достоинство комбинации OsO 4 и NaIO 4 заключается в том, что она позволяет остановить окисление на стадии альдегида. Тетраоксид осмия присоединяется к двойной связи алкена с образованием осмата, который окисляется периодатом натрия до карбонильных соединений с регенерацией четырехокиси осмия.

Вместо тетраоксида осмия можно использовать и тетраоксид рутения RuO 4 . Окислительная деструкция алкенов по Лемье-Джонсону приводит к тем же продуктам, что и озонолиз с восстановительным расщеплением озонидов.

В терминах, характерных для современной органической химии, это означает, что комбинация OsO 4 -NaIO 4 представляет собой синтетический эквивалент реакции озонолиза алкенов с последующим восстановительным расщеплением. Аналогично, окисление алкенов смесью перманганата и периодата - это синтетический эквивалент озонолиза с окислительным разложением озонидов.
Таким образом, окисление алкенов - это не только совокупность препаративных методов получения спиртов, эпоксидов, диолов, альдегидов, кетонов и карбоновых кислот, это также один из возможных путей установления структуры исходного алкена. Так, по результату, окислительной деструкции алкена можно определить положение двойной связи в молекуле, тогда как стереохимический результат син- или анти- гидроксилирования алкена позволяет сделать вывод о его геометрии.
Санкт-Петербургский Государственный Технологический Институт
(Технический Университет)
Кафедра органической химии Факультет 4
Группа 476
Курсовая работа
Окисление алкенов
Студентка………………………………………Рытина А.И.
Преподаватель………………………………... Питерская Ю.Л.
Санкт-Петербург
Введение
1.Эпоксидирование (реакция Н.А. Прилежаева,1909 г.)
2.Гидроксилирование
2.1анти -Гидроксилирование
2.2син -Гидроксилирование
3.Окислительное расщепление алкенов
4.Озонолиз
5.Окисление алкенов в присутствии солей палладия
Заключение
Список использованных источников
Введение
Окисление - одно из наиболее важных и распространенных превращений органических соединений.
Под окислением в органической химии понимают процессы, приводящие к обеднению соединения водородом или обогащению его кислородом. При этом происходит отнятие от молекулы электронов. Соответственно, под восстановлением понимают отрыв от органической молекулы кислорода или присоединение к ней водорода.
В окислительно-восстановительных реакциях окислителями являются соединения, обладающие большим сродством к электрону (электрофилы), а восстановителями – соединения, имеющие склонность к отдаче электронов (нуклеофилы). Легкость окисления соединения возрастает вместе с ростом его нуклеофильности.
При окислении органических соединений, как правило, полной передачи электронов и соответственно изменения валентности атомов углерода не происходит. Поэтому понятие степени окисления – условного заряда атома в молекуле, вычисленного, исходя из предположения, что молекула состоит только из ионов – носит лишь условный, формальный характер.
При составлении уравнений окислительно-восстановительных реакций необходимо определить восстановитель, окислитель и число отдаваемых и принимаемых электронов. Как правило, коэффициенты подбирают, используя метод электронно-ионного баланса (метод полуреакций).
В этом методе рассматривают переход электронов от одних атомов или ионов к другим с учетом характера среды (кислая, щелочная или нейтральная), в которой протекает реакция. Для уравнивания числа атомов кислорода и водорода вводят или молекулы воды и протоны (если среда кислая), или молекулы воды и гидроксид-ионы (если среда щелочная).
Таким образом, при написании полуреакций восстановления и окисления нужно исходить из состава ионов, действительно имеющихся в растворе. Вещества малодиссоциирующие, плохо растворимые или выделяющиеся в виде газа следует писать в молекулярной форме.
В качестве примера рассмотрим процесс окисления этилена разбавленным водным раствором перманганата калия (реакция Вагнера). В ходе данной реакции этилен окисляется до этиленгликоля, а перманганат калия восстанавливается до диоксида марганца. По месту двойной связи присоединяются два гидроксила 1:
3С 2 H 4 + 2KMnO 4 +4H 2 O→ 3C 2 H 6 O 2 + 2MnO 2 +2KOH
Полуреакция восстановления: MnO 4 ¯ + 2H 2 O + 3 e → MnO 2 + 4OH ¯ 2
Полуреакция окисления: С 2 H 4 + 2OH − − 2 e → C 2 H 6 O 2 3
Окончательно имеем в ионном виде:
2MnO 4 ¯ + 4H 2 O + 3C 2 H 4 + 6OH ¯ → 2MnO 2 + 8OH ¯ + 3C 2 H 6 O 2
После проведения необходимых сокращений подобных членов, записываем уравнение в молекулярном виде:
3C 2 H 4 + 2KMnO 4 + 4 H 2 O = 3C 2 H 6 O 2 + 2MnO 2 + 2KOH.
Характеристика некоторых окислителей
Кислород
Кислород воздуха находит широкое применение в технологических процессах, так как является наиболее дешевым окислителем. Но окисление кислородом воздуха сопряжено с трудностями, связанными с контролем процесса, который протекает в различных направлениях. Окисление обычно проводят при высокой температуре в присутствии катализаторов.
Озон
Озон O 3 применяют для получения альдегидов и кетонов, если их затруднительно получить другими способами. Чаще всего озон применяют для установления структуры ненасыщенных соединений. Получают озон при действии тихого электрического разряда на кислород. Одним из существенных достоинств озонирования, по сравнению с хлорированием, является отсутствие токсинов после обработки 2 .
Перманганат калия
Перманганат калия – наиболее часто применяемый окислитель. Реактив растворим в воде (6.0% при 20ºС), а также в метаноле, ацетоне и уксусной кислоте. Для окисления применяют водные (иногда ацетоновые) растворы KMnO 4 в нейтральной, кислой или щелочной среде. При проведении процесса в нейтральной среде в реакционную массу добавляют соли магния, алюминия или пропускают углекислый газ для нейтрализации выделяющегося во время реакции гидроксида калия. Реакцию окисления KMnO 4 в кислой среде чаще всего ведут в присутствии серной кислоты. Щелочную среду при окислении создает образующийся во время реакции KOH, либо его изначально добавляют в реакционную массу. В слабощелочной и нейтральной средах KMnO 4 окисляет по уравнению:
KMnO 4 + 3 e + 2H 2 O = K + + MnO 2 + 4OH ¯
в кислой среде:
KMnO 4 + 5 e + 8H + = K + + Mn 2+ + 4H 2 O 3
Перманганат калия используется для получения 1,2-диолов из алкенов, при окислении первичных спиртов, альдегидов и алкиларенов до карбоновых кислот, а также для окислительного расщепления углеродного скелета по кратным связям.
На практике обычно используется довольно большой избыток (более чем 100%) KMnO 4 . Это объясняется тем, что в обычных условиях KMnO 4 частично разлагается на диоксид марганца с выделением O 2 . Разлагается концентрированной H 2 SO 4 при нагревании в присутствии восстановителей со взрывом; смеси калия перманганата с органическими веществами также взрывчаты 4 .
Надкислоты
Перуксусную и пермуравьиную кислоты получают реакцией 25-90%-ного пероксида водорода с соответствующей карбоновой кислотой по следующей реакции:
RCOOH + H 2 O 2 = RCOOOH + H 2 O
В случае уксусной кислоты это равновесие устанавливается относительно медленно, и для ускорения образования перкислоты обычно в качестве катализатора добавляют серную кислоту. Муравьиная кислота достаточно сильна сама по себе для того, чтобы обеспечить быстрое установление равновесия.
Пертрифторуксусная кислота, получаемая в смеси с трифторуксусной кислотой реакцией трифторуксусного ангидрида с 90%-ным пероксидом водорода, еще более сильный окислитель. Аналогичным образом из уксусного ангидрида и пероксида водорода можно получить перуксусную кислоту.
Особой популярностью пользуется твердая м -хлорпербензойная кислота, поскольку она относительно безопасна в обращении, достаточно стабильна и может храниться длительное время.
Окисление происходит за счет выделяющегося атома кислорода:
RCOOOH = RCOOH + [O]
Надкислоты применяют для получения эпоксидов из алкенов, а также лактонов из алициклических кетонов.
Пероксид водорода
Пероксид водорода – бесцветная жидкость,cмешивается с водой, этанолом и диэтиловым эфиром. 30%-ный раствор H 2 O 2 называется пергидролем. Высококонцентрированный препарат может реагировать с органическими веществами со взрывом. При хранении разлагается на кислород и воду. Стойкость пероксида водорода возрастает с разбавлением. Для окисления применяют водные растворы различной концентрации (от 3 до 90%) в нейтральной, кислой или щелочной средах.
H 2 O 2 = H 2 O + [O]
Действием этого реагента на α,β-непредельные карбонильные соединения в щелочной среде получают соответствующие эпоксиальдегиды и кетоны, окислением карбоновых кислот в кислой среде синтезируют надкислоты. 30%-ный раствор H 2 O 2 в уксусной кислоте окисляет алкены в 1,2-диолы. Пероксид водорода применяют: для получения органических и неорганических пероксидов, пербората и перкарбоната Na; как окислитель в ракетных топливах; при получении эпоксидов, гидрохинона, пирокатехина, этиленгликоля, глицерина, ускорителей вулканизации группы тиурама и др.; для отбеливания масел, жиров, меха, кожи, текстильных материалов, бумаги; для очистки германиевых и кремниевых полупроводниковых материалов; как дезинфицирующее средство для обезвреживания бытовых и индустриальных сточных вод; в медицине; как источник О 2 в подводных лодках; Н 2 О 2 входит в состав реактива Фентона (Fe 2 + + Н 2 О 2), который используют как источник свободных радикалов ОН в органическом синтезе 5 .
Тетраоксиды рутения и осмия
Тетраоксид осмия OsO 4 – порошок от белого до бледно-желтого цвета с т. пл. 40.6ºС; т. кип. 131.2ºС. Возгоняется уже при комнатной температуре, растворим в воде (7.47 г в 100 мл при 25ºС), ССl 4 (250 г в 100 г растворителя при 20ºС). В присутствии органических соединений чернеет вследствие восстановления до OsO 2 6 .
RuO 4 представляет собой золотисто-желтые призмы с т. пл. 25.4ºС, заметно возгоняется при комнатной температуре. Умеренно растворим в воде (2.03 г в 100 мл при 20ºС), очень хорошо растворим в CCl 4 . Более сильный окислитель, чем OsO 4 . Выше 100ºС взрывается. Как и тетраоксид осмия обладает большой токсичностью и высокой стоимостью.
Данные окислители применяются для окисления алкенов в α-гликоли в мягких условиях.
Диоксираны
Наиболее часто используются диметилдиоксиран и метил(трифторметил)-диоксиран.

Диоксираны чаще всего получают in situ из соответствующих кетонов и KHSO 5 (или K 2 SO 4 ·KHSO 4 ·2KHSO 5 (оксона)) в слабощелочной среде 7:
Диоксираны отличаются высокой реакционной способностью, сочетающейся с хорошей селективностью, и используются для окисления неактивированных С–Н-связей в алканах, при получении эпоксидов из алкенов, для окисления аминов, оксимов, сульфидов, сульфоксидов и др.
Окисление алкенов может проходить в нескольких направлениях:
1)с сохранением углеродного скелета молекулы; так протекают эпоксидирование и гидроксилирование двойной связи,приводящее к образаванию вицинальных транс- или цис-гликолей.
2)с разрывом двойной связи; так идут озонолиз и исчерпывающее окисление алкенов,приводящее к образованию различного рода карбонильных соединений и карбоновых кислот.
В зависимости от типа окисления применяют различные окислители
1. Эпоксидирование (реакция Н.А. Прилежаева, 1909 г)
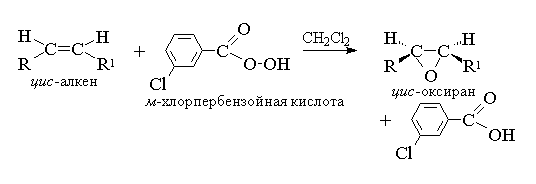
Ациклические и циклические алкены при взаимодействии с перкислотами (надкислотами) RCOOOH в неполярной, индифферентной среде образуют эпоксиды (оксираны), поэтому сама реакция носит название реакции эпоксидирования.

Согласно современной номенклатуре ИЮПАК - трехчленный цикл с одним атомом кислорода носит название оксиран.
Эпоксидирование алкенов следует рассматривать как синхронный, согласованный процесс, в котором не участвуют ионные интермедиаты типа гидроксильного катиона ОН+. Другими словами, эпоксидирование алкенов представляет собой процесс син -присоединения одного атома кислорода по двойной связи с полным сохранением конфигурации заместителей при двойной связи. 8

Для эпоксидирования был предложен механизм, характерный для согласованных процессов.

Т. к. атака двойной связи атомом кислорода надкислоты равновероятна с обеих сторон плоскости двойной связи, образующиеся оксираны представляют собой либо мезо -формы, либо смеси энантиомеров. В качестве эпоксидирующих агентов используются следующие перкислоты: пербензойная, м -хлорпербензойная, моноперфталевая, перуксусная, трифторперуксусная и пермуравьиная. Перкислоты ароматического ряда применяют в виде индивидуальных реагентов, тогда как перкислоты алифатического ряда - СН 3 СО 3 Н, CF 3 CO 3 H и НСО 3 Н не выделяют в индивидуальном виде, а используют после их образования при взаимодействии 30% или 90%-ного пероксида водорода и соответствующей карбоновой кислоты. Пербензойную и м -хлорпербензойную кислоты получают окислением соответственно бензойной и м -хлорбензойной кислот 70%-ной перекисью водорода в растворе метансульфокислоты или из хлорангидридов этих кислот и перекиси водорода.


Моноперфталевую кислоту получают подобным методом из фталевого ангидрида и 30%-ной перекиси водорода.

В настоящее время значительное применение находит монопероксифталевая кислота в виде Mg-соли.Этот реагент можно применять в смесях органических растворителей с водой 9 .
Первоначально для получения оксиранов (эпоксидов) использовались пербензойная или моноперфталевая кислоты 10:

В настоящее время для эпоксидирования чаще всего используют м -хлорпербензойную кислоту. В отличие от других перкислот она стабильна при хранении в течение длительного времени (до 1 года) и абсолютно безопасна при обращении. Выходы оксиранов, полученных при окислении ациклических и циклических алкенов м -хлорпербензойной кислотой в растворе хлористого метилена, хлороформа или диоксана, обычно довольно высоки 11 .



Перкислоты часто генерируют прямо в реакционной смеси из 90% перекиси водорода и карбоновой кислоты в хлористом метилене.

Алкены с двойной связью, сопряженной с карбонильной группой или другим акцепторным заместителем, малоактивны и для их окисления лучше использовать более сильные окислители, такие как трифторперуксусная кислота, получаемая из ангидрида трифторуксусной кислоты и 90%-ной перекиси водорода в хлористом метилене. Пероксикарбоксимидовые кислоты RC(NH)OOH являются нестойкими промежуточными продуктами, образующимися в реакциях нитрилов со щелочными растворами пероксида водорода 12:
Простейший оксиран - окись этилена получают в промышленности окислением этилена кислородом в присутствии серебра, как катализатора 13 .

2.Гидроксилирование
Известен ряд окислительных реагентов, с помощью которых в мягких условиях возможно присоединение двух гидроксильных групп к алкенам.
Реакция гидроксилирования алкенов, протекающая под действием холодного раствора перманганата калия и сопровождаемая его обецвечиванием, известна как реакция Вагнера(1888 г.). Она имеет в настоящее время незначительное синтетическое применение, поскольку сопровождается образованием значительного числа побочных продуктов. Однако это реакция может применяться при изучении строения органического соединения как качественная проба на двойную связь. Гидроксилирование циклогексена действием водного раствора перманганата калия на холоду впервые провел В.В. Марковников. В настоящее время для гидроксилирования алкенов чаще всего применяют оксид осмия(VIII)(реакция Криге,1936г.) 14 .
Четырехокись осмия OsO 4 представляет собой бесцветное кристаллическое вещество,плавящееся при 40 о С и легко растворимое в эфире. При окислении циклогексена эфирным раствором этого реагента выделяется черный осадок, который представляет собой циклический эфир осмиевой кислоты I 15:
Эфир образуется в результате одновременного раскрытия углерод-углеродной двойной связи алкена и двух двойных связей окиси металла. Затем этот эфир гидролизуют, используя в качестве катализатора сульфит натрия. Продукт гидролиза представляет собой цис-циклогександиол-1,2(II),у которого гидроксилы расположены в β-области,т.е. спереди, а водороды-в α-области,т.е. сзади.
2.1 анти -Гидроксилирование
Трехчленное кольцо оксиранов легко раскрывается под действием самых разнообразных нуклеофильных реагентов. Гидролиз оксиранов катализируется как кислотами, так и основаниями. В обоих случаях образуются вицинальные диолы, т. е. гликоли. При кислотном катализе в первой стадии происходит протонирование атома кислорода оксирана с образованием циклического оксониевого катиона, который раскрывается в результате нуклеофильной атаки молекулы воды 16:

Ключевой стадией в раскрытии кольца, определяющей скорость всего процесса, является нуклеофильная атака водой на протонированную форму оксирана. С точки зрения механизма этот процесс аналогичен раскрытию бромониевого иона при нуклеофильной атаке бромид-иона или другого нуклеофильного агента. С этих позиций стереохимическим результатом должно быть образование транс -гликолей при расщеплении циклических эпоксидов. Действительно, при кислотно-катализируемом гидролизе циклогексеноксида или циклопентеноксида образуются исключительно транс -1,2-диолы.
Таким образом, двухстадийный процесс эпоксидирования алкена с последующим кислотным гидролизом эпоксида суммарно соответствует реакции анти -гидроксилирования алкенов.
Обе стадии анти -гидроксилирования алкенов можно совместить, если алкен обрабатывать водной 30-70%-ной перекисью водорода в муравьиной или трифторуксусной кислоте. Обе эти кислоты являются достаточно сильными для того, чтобы вызвать раскрытие оксиранового цикла.

Раскрытие оксиранового кольца, катализируемое основанием, также приводит к образованию циклических транс -гликолей.
Следовательно, двухстадийный процесс эпоксидирования алкенов с последующим щелочным гидролизом эпоксидов также является реакцией анти -гидроксилирования алкенов.
2.2 син -Гидроксилирование
Некоторые соли и оксиды переходных металлов в высших степенях окисления являются эффективными реагентами син -гидроксилирования двойной связи алкена, когда обе гидроксильные группы присоединяются с одной и той же стороны двойной связи. Окисление алкенов перманганатом калия - один из старейших методов син -гидроксилирования двойной связи - продолжает широко использоваться, несмотря на свойственные ему ограничения. Цис -1,2-циклогександиол был впервые получен В.В. Марковниковым в 1878 году гидроксилированием циклогексена водным раствором перманганата калия при 0 0 С.

Этот метод в дальнейшем получил развитие в работах русского ученого Е.Е. Вагнера, поэтому син -гидроксилирование алкенов под действием водного раствора перманганата калия носит название реакции Вагнера. Перманганат калия является сильным окислителем, способным не только гидроксилировать двойную связь, но и расщеплять образующийся вицинальный диол. Для того, чтобы по возможности избежать дальнейшего расщепления гликолей, необходимо тщательно контролировать условия реакции. Выходы гликолей при этом обычно невелики (30-60%). Наилучшие результаты достигаются при гидроксилировании алкенов в слабощелочной среде (рН~8 9) при 0-5 0 С разбавленным 1%-ным водным раствором KMnO 4 17 .


Первоначально при окислении алкенов перманганатом калия образуется циклический эфир марганцевой кислоты, который немедленно гидролизуется до вицинального диола.

Циклический эфир марганцевой кислоты как интермедиат не был выделен, однако его образование следует из экспериментов с меченым 18 О перманганатом калия: оба атома кислорода в гликоле оказываются мечеными при окислении алкена KMn 18 O 4 . Это означает, что оба атома кислорода переходят от окислителя, а не из растворителя - воды, что находится в хорошем соответствии с предлагаемым механизмом.
Другой метод син -гидроксилирования алкенов под действием оксида осмия (VIII) OsO 4 был предложен Р. Криге в 1936 году. Тетраоксид осмия представляет собой бесцветное, летучее, кристаллическое вещество, хорошо растворимое в эфире, диоксане, пиридине и др. органических растворителях. При взаимодействии тетраоксида осмия с алкенами в эфире или диоксане образуется черный осадок циклического эфира осмиевой кислоты - осмат, который легко может быть изолирован в индивидуальном виде. Присоединение OsO 4 к двойной связи заметно ускоряется в растворе в пиридине. Разложение осматов до вицинальных гликолей достигается действием водного раствора гидросульфита натрия или сероводородом.

Выходы продуктов син -гидроксилирования алкенов в этом методе значительно выше, чем при использовании перманганата в качестве окислителя. Важным достоинством метода Криге является отсутствие продуктов окислительного расщепления алкенов, характерного для перманганатного окисления 18 .
 19
19

Тетраоксид осмия очень дорогой и труднодоступный реагент, к тому же он токсичен. Поэтому оксид осмия (VIII) используется при синтезе малых количеств трудно доступных веществ с целью получения наиболее высокого выхода диола. С целью упрощения син -гидроксилирования алкенов под действием OsO 4 была разработана методика, позволяющая использовать лишь каталитические количества этого реагента. Гидроксилирование алкенов осуществляется с помощью перекиси водорода в присутствии OsO 4 , например:

В заключение можно привести стереохимические отношения между алкеном цис - или транс -конфигурации и конфигурацией образующегося вицинального диола, который может быть цис - или транс -изомером, эритро - или трео -формой, мезо - или D,L -формой в зависимости от заместителей в алкене 20:

Аналогичные стереохимические отношения наблюдаются и в других реакциях син - или анти -присоединения по кратной связи водорода, галогенводородов, воды, галогенов, гидридов бора и др. реагентов.
3. Окислительное расщепление алкенов
При окислении алкенов щелочным водным раствором перманганата калия при нагревании или раствором KMnO 4 в водной серной кислоте, а также при окислении алкенов раствором оксида хрома (VI) CrO 3 в уксусной кислоте или дихроматом калия и серной кислотой первоначально образующийся гликоль подвергается окислительной деструкции. Конечным результатом является расщепление углеродного скелета по месту двойной связи и образование в качестве конечных продуктов кетонов и (или) карбоновых кислот в зависимости от заместителей при двойной связи. Если оба атома углерода при двойной связи содержат только по одной алкильной группе, конечным продуктом исчерпывающего окисления будет смесь карбоновых кислот, тетразамещенный при двойной связи алкен окисляется до двух кетонов. Однозамещанные алкены с концевой двойной связью расщепляются до карбоновой кислоты и углекислого газа 21 .

 22
22
Из-за невысоких выходов карбоновых кислот и кетонов, реакции исчерпывающего окисления алкенов в классическом варианте не нашли широкого применения и ранее использовались, в основном, для установления строения исходного алкена по продуктам деструктивного окисления. В настоящее время окисление алкенов (R-CH=CH-R и R-CH=CH 2) до карбоновых кислот (RCOOH) с помощью перманганата или дихромата калия проводят в условиях межфазного катализа. Выходы карбоновых кислот при этом превышают 90%.

4.Озонолиз алкенов
Реакция алкенов с озоном является наиболее важным методом окислительного расщепления алкенов по двойной связи. В течение многих десятилетий эта реакция служила основным методом определения строения исходного углеводорода, а также находила применение в синтезе разнообразных карбонильных соединений. Реакция алкена с озоном проводится пропусканием тока ~5%-ной смеси озона и кислорода в раствор алкена в хлористом метилене или этилацетате при -80 0 -100 0 С. Окончание реакции контролируется пробой на свободный озон с иодидом калия. Механизм этой своеобразной и сложной реакции установлен главным образом благодаря работам Криге. Первым продуктом 1,3-диполярного циклоприсоединения к двойной связи является так называемый мольозонид (1,2,3-триоксолан). Этот продукт нестабилен и далее самопроизвольно разлагается с раскрытием цикла и образованием в качестве конечного продукта нормального озонида (1,2,4-триоксолана) 23 .
 24
24
В настоящее время общепризнано, что превращение мольозонида в обычный озонид происходит по механизму расщепления - рекомбинации. Мольозонид претерпевает самопроизвольное раскрытие нестабильного 1,2,3-триоксоланового цикла с образованием карбонильного соединения и биполярного иона, которые далее реагируют между собой также по схеме 1,3-диполярного циклоприсоединения.

Приведенная схема перегруппировки мольозонида в нормальный озонид подтверждается тем, что если до полного образования озонида в реакционной смеси присутствует в качестве "перехватчика" биполярного иона другое карбонильное соединение, то образуется так называемый "смешанный озонид". Так, например, при озонилизе цис -стильбена в присутствии бензальдегида, меченного изотопом 18 О, метка входит в состав эфирного, а не перекисного мостика озонида:
Этот результат хорошо согласуется с образованием смешанного озонида при рекомбинации биполярного иона с меченным бензальдегидом:

Озониды представляют собой очень нестабильные соединения, разлагающиеся со взрывом. Их не выделяют в индивидуальном виде, а расщепляют при действии самых разнообразных регентов. Следует различать восстановительное и окислительное расщепление. При гидролизе озониды медленно расщепляются на карбонильные соединения и перекись водорода. Перекись водорода окисляет альдегиды до карбоновых кислот. Это так называемое окислительное разложение озонидов:

Таким образом, при окислительном разложении озонидов образуются карбоновые кислоты и (или) кетоны в зависимости от строения исходного алкена. В качестве окислителей можно использовать кислород воздуха, перекись водорода, перкислоты или гидроокись серебра. Наиболее часто в синтетической практике для этой цели используют перекись водорода в уксусной или муравьиной кислоте, а также перекись водорода в щелочной среде.


На практике метод окислительного разложения озонидов используется, в основном, для получения карбоновых кислот.
Более важное значение имеет восстановительное расщепление озонидов. В качестве восстановителей наиболее часто используются цинк и уксусная кислота, трифенилфосфин или диметилсульфид. В этом случае конечными продуктами озонолиза оказываются альдегиды или кетоны в зависимости от строения исходного алкена.


 25
25
Из приведенных выше примеров видно, что тетразамещенный при двойной связи алкен при озонолизе и последующем восстановительном разложении озонида образует два кетона, тогда как тризамещенный алкен дает кетон и альдегид. Дизамещенный симметричный алкен при озонолизе образует два альдегида, а алкены с концевой связью - альдегид и формальдегид.
Интересной модификацией озонолиза является метод, где в качестве восстановителя озонида используется боргидрид натрия, В этом случае конечными продуктами реакции оказываются первичные или вторичные спирты, образующиеся при восстановлении соответственно альдегидов и кстонов 26 .


Озонолиз алкенов - это сложный, трудоемкий и взрывоопасный процесс, требующий применения специальной аппаратуры. По этой причине были разработаны другие методы окислительного расщепления алкенов до карбонильных соединений и карбоновых кислот, которые с успехом заменяют реакцию озонолиза в синтетической практике.
Один из современных препаративных методов окислительной деструкции алкенов был предложен в 1955 г Р. Лемье. В основе этого метода лежит гидроксилирование алкенов с помощью перманганата калия с последующим расщеплением вицинального гликоля периодатом натрия NaIO 4 при рН ~ 7 8. Периодат сам по себе не взаимодействует с алкеном. Продуктами этого двухстадийного окислительного расщепления являются кетоны или карбоновые кислоты, поскольку альдегиды в этих условиях также окисляются до карбоновых кислот. В методе Лемье не возникает трудоемкой проблемы отделения одного из продуктов реакции, - двуокиси марганца, так как и двуокись, и манганат вновь окисляются периодатом до перманганат-иона. Это позволяет использовать только каталитические количества перманганата калия. Ниже приведены некоторые типичные примеры окислительного расщепления алкенов по методу Лемье.

Цитронеллол - спирт, входящий в состав розового масла, масла герани и лимона, - окисляется смесью перманганата калия и периодата натрия в водном ацетоне при 5 10 0 С до 6-гидрокси-4-метилгексанкарбоновой кислоты с количественным выходом.

В другой разновидности этого метода вместо перманганата калия используют каталитические количества тетраоксида осмия (Лемье, Джонсон 1956 г). Особое достоинство комбинации OsO 4 и NaIO 4 заключается в том, что она позволяет остановить окисление на стадии альдегида. Тетраоксид осмия присоединяется к двойной связи алкена с образованием осмата, который окисляется периодатом натрия до карбонильных соединений с регенерацией четырехокиси осмия.

Вместо тетраоксида осмия можно использовать и тетраоксид рутения RuO 4 . Окислительная деструкция алкенов по Лемье-Джонсону приводит к тем же продуктам, что и озонолиз с восстановительным расщеплением озонидов.

В терминах, характерных для современной органической химии, это означает, что комбинация OsO 4 -NaIO 4 представляет собой синтетический эквивалент реакции озонолиза алкенов с последующим восстановительным расщеплением. Аналогично, окисление алкенов смесью перманганата и периодата - это синтетический эквивалент озонолиза с окислительным разложением озонидов.
Таким образом, окисление алкенов - это не только совокупность препаративных методов получения спиртов, эпоксидов, диолов, альдегидов, кетонов и карбоновых кислот, это также один из возможных путей установления структуры исходного алкена. Так, по результату, окислительной деструкции алкена можно определить положение двойной связи в молекуле, тогда как стереохимический результат син- или анти- гидроксилирования алкена позволяет сделать вывод о его геометрии.
5.Окисление алкенов в присутствии солей палладия
Окислению алкенов в карбонильные соединения с помощью хлорида палладия посвящен значительное количество исследований в основном благодаря важному значению для промышленности реакции получения ацетальдегида из этилена (Вакер-процесс). В ходе окисления хлорид палладия восстанавливается в палладий, и такой процесс представлял бы ограниченный интерес, если бы не то обстоятельство, что дорогой хлорид палладия можно использовать в каталитических количествах в присутствии второго окислителя, чаще всего хлорида меди(II),который окисляет палладий в палладий(II),сам восстанавливаясь при этом до меди (I).Реокисление меди(I) в медь(II) можно осуществить атмосферным кислородом, так что суммарный процесс весьма привлекателен в качестве промышленного метода окисления 27 .
Этилен легко окисляется до ацетальдегида 28:
Предполагаемый механизм 29:
Реакция идет в кислой среде, не сопровождается изменением числа атомов углерода в молекуле этилена и является в настоящее время основным источником ацетальдегида в промышленности.
Окисление гомологов этилена в тех же условиях идет по наименее гидрогенезированному атому углерода двойной связи с образованием кетонов. В частности, при окислении пропена получают ацетон, а при окислении циклогексена-циклогексанон.
Заключение
Реакция окисления является важной группой реакций двойной связи. Вообще, реакции окисления в органической химии занимают особое место. При рассмотрении этих реакций приходится учитывать не только природу окисляемого органического соединения, но и природу окислителя, наличие или отсутствие катализатора, среды, в которой протекает реакция и т.д.
Поэтому приходится очень часто запоминать окислители и условия их применения для получения нужного продукта окисления. Например, реакция окисления алкенов разбавленным раствором перманганата калия приводит к образованию диолов (гликолей), в то время как концентрированный раствор перманганата калия разрушает молекулу алкена по двойной связи с образованием кислородсодержащих продуктов. Таким образом, один и тот же окислитель в различных средах дает разные продукты окисления.
Среди реакций окисления наиболее важной считается рассмотренная нами реакция озонирования, которая позволяет по конечным продуктам установить структуру исходного алкена. В данной работе были рассмотрены основные реакции окисления алкенов и катализаторы, которые используются в ходе окислительных процессов.
Список использованных источников:
1)Реутов О. А. Органическая химия.В 4-х частях.Ч. 1.-М.:БИНОМ. Лаборатория знаний,2004.-567с.
2)Реми Г. Курс неорганической химии. Т. 1. М.: Изд-во иностранной литературы, 1963. – 920 с.
3)Казаков Д.В., Волошин А.И., Казаков В.П., Шерещовец В.В., Кабальнова Н.Н. Химия и хемилюминесценция диоксиранов. М.: Наука, 1999
4) Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т / В.Ф.Травень. - М.: ИКЦ «Академкнига», 2004. - Т. 1
5)Хейнс А. Методы окисления органических соединений. Алканы, алкены, алкины и арены. М.: Мир, 1988.
6)Моррисон Р., Бойд Р. "Органическая химия" М.:Мир, 1974
7)Шабаров Ю.С. "Органическая химия" ч.1 М.:Химия 1994
8)Петров А.А.,Бальян Х. В.,Трощенко А.Т. Органическая химия:Учебник для вузов.-5-е изд.,перераб. И доп.-СПб.: «Иван Федоров»,2002.-624 с.
9)Л. Физер,М. Физер Органическая химия.Углубленный курс.Том 1-М., «Химия»,1969 г.,688 с.
10)Плотников В.Ф.,Питерская Ю.Л. Алкены.Учебное пособие.-СПб:СПбГТИ(ТУ)-2003.-20 с.
11) Нейланд О. Я. Глава II. Алкены // Органическая химия: Учеб. для хим. вузов. - М.: «Высшая школа», 1990.
12) Марч Д. Органическая химия: В 4-х т./Пер. с англ. – М.: Мир, 1987. – Т. 4. 470 с.
13) Терней А. Современная органическая химия: В 2-х т. – М.: Мир, 1981.
14) Гауптман З., Грефе Ю., Ремане Х. "Органическая химия" М.:Химия, 1979
15) Дрюк В.Г., Малиновский М.С. "Курс органической химии" Киев:Вища школа 1987
16) http://www.chemistry.ssu.samara.ru/chem2/u442.htm
17) http://www.xumuk.ru/
1 Петров А.А.,Бальян Х.В.,Трощенко А.Т. Органическая химия: Учебник для вузов,с.85
26 26 ТравеньРеферат >> Химия
CH2 + 3O2 2CO2 + 2H2O b) При окислении алкенов разбавленным раствором перманганата калия образуются... на двойную связь. c) При жестком окислении алкенов кипящим раствором перманганата калия в кислой...
Теория общей химии с элементами методики преподавания
Шпаргалка >> ХимияСпособ получения карбонильных соединений - окисление спиртов. В качестве окислителя можно... выход~90%) 5. Окисление алкенов . Альдегиды и кетоны получают окислением углеводородов ряда этилена... не гидратируются. 7. Реакции окисления . Альдегиды и кетоны по- ...
Оксираны (эпоксиды)
Реферат >> Химия...) 3,4-эпокси-1-бутен Методы получения оксиранов Окисление алкенов надкислотами Наиболее широко используемым методом... 1,2-эпоксицикло- м-хлорбензойная кислота гексан кислота Окисление алкенов органическими надкислотами сопровождается присоединением кислорода...
Общие сведения о спиртах. Полиолы
Реферат >> Химия...) могут быть получены окислением алкенов перманганатом калия или тетрокисдом... . Опишите механизм реакции Окисление полиолов Окисление этиленгликоля протекает различно, ... механизмов: (м 21) Периодатное окисление глицерина приводит к образованию формальдегида...
4.5. Окисление алкенов
Реакции окисления алкенов целесообразно подразделить на две большие группы: реакции, в которых сохраняется углеродный скелет и реакции окислительной деструкции углеродного скелета молекулы по двойной связи. К первой группе реакций относятся эпоксидирование, а также гидроксилирование, приводящее к образованию вицинальных диолов (гликолей). В случае циклических алкенов при гидроксилировании образуются вицинальные транс - или цис -диолы. Другая группа включает озонолиз и реакции исчерпывающего окисления алкенов, приводящие к образованию различного рода карбонильных соединений и карбоновых кислот.
4.5.а. Реакции окисления алкенов с сохранением углеродного скелета
1. Эпоксидирование (реакция Н.А. Прилежаева, 1909 г)
Ациклические и циклические алкены при взаимодействии с перкислотами (надкислотами) RCOOOH в неполярной, индифферентной среде образуют эпоксиды (оксираны), поэтому сама реакция носит название реакции эпоксидирования.
Согласно современной номенклатуре ИЮПАК - трехчленный цикл с одним атомом кислорода носит название оксиран.
Эпоксидирование алкенов следует рассматривать как синхронный, согласованный процесс, в котором не участвуют ионные интермедиаты типа гидроксильного катиона ОН+ . Другими словами, эпоксидирование алкенов представляет собой процесс син -присоединения одного атома кислорода по двойной связи с полным сохранением конфигурации заместителей при двойной связи.

Для эпоксидирования был предложен механизм, характерный для согласованных процессов.

Т. к. атака двойной связи атомом кислорода надкислоты равновероятна с обеих сторон плоскости двойной связи, образующиеся оксираны представляют собой либо мезо -формы, либо смеси энантиомеров. В качестве эпоксидирующих агентов используются следующие перкислоты: пербензойная, м -хлорпербензойная, моноперфталевая, перуксусная, трифторперуксусная и пермуравьиная. Перкислоты ароматического ряда применяют в виде индивидуальных реагентов, тогда как перкислоты алифатического ряда - СН 3 СО 3 Н, CF 3 CO 3 H и НСО 3 Н не выделяют в индивидуальном виде, а используют после их образования при взаимодействии 30% или 90%-ного пероксида водорода и соответствующей карбоновой кислоты. Пербензойную и м -хлорпербензойную кислоты получают окислением соответственно бензойной и м -хлорбензойной кислот 70%-ной перекисью водорода в растворе метансульфокислоты или из хлорангидридов этих кислот и перекиси водорода.


Моноперфталевую кислоту получают подобным методом из фталевого ангидрида и 30%-ной перекиси водорода.

Первоначально для получения оксиранов (эпоксидов) использовались пербензойная или моноперфталевая кислоты:

В настоящее время для эпоксидирования чаще всего используют м -хлорпербензойную кислоту. В отличие от других перкислот она стабильна при хранении в течение длительного времени (до 1 года) и абсолютно безопасна при обращении. Выходы оксиранов, полученных при окислении ациклических и циклических алкенов м -хлорпербензойной кислотой в растворе хлористого метилена, хлороформа или диоксана, обычно довольно высоки.



Перкислоты часто генерируют прямо в реакционной смеси из 90% перекиси водорода и карбоновой кислоты в хлористом метилене.


Алкены с двойной связью, сопряженной с карбонильной группой или другим акцепторным заместителем, малоактивны и для их окисления лучше использовать более сильные окислители, такие как трифторперуксусная кислота, получаемая из ангидрида трифторуксусной кислоты и 90%-ной перекиси водорода в хлористом метилене. Простейший оксиран - окись этилена получают в промышленности окислением этилена кислородом в присутствии серебра, как катализатора.

2. анти -Гидроксилирование
Трехчленное кольцо оксиранов легко раскрывается под действием самых разнообразных нуклеофильных реагентов. Эти реакции подробно будут обсуждаться в разделе, посвященном ациклическим и циклическим простым эфирам. Здесь же будет рассматриваться только гидролиз оксиранов. Гидролиз оксиранов катализируется как кислотами, так и основаниями. В обоих случаях образуются вицинальные диолы, т. е. гликоли. При кислотном катализе в первой стадии происходит протонирование атома кислорода оксирана с образованием циклического оксониевого катиона, который раскрывается в результате нуклеофильной атаки молекулы воды:

Ключевой стадией в раскрытии кольца, определяющей скорость всего процесса, является нуклеофильная атака водой на протонированную форму оксирана. С точки зрения механизма этот процесс аналогичен раскрытию бромониевого иона при нуклеофильной атаке бромид-иона или другого нуклеофильного агента. С этих позиций стереохимическим результатом должно быть образование транс -гликолей при расщеплении циклических эпоксидов. Действительно, при кислотно-катализируемом гидролизе циклогексеноксида или циклопентеноксида образуются исключительно транс -1,2-диолы.

Таким образом, двухстадийный процесс эпоксидирования алкена с последующим кислотным гидролизом эпоксида суммарно соответствует реакции анти -гидроксилирования алкенов.
Обе стадии анти -гидроксилирования алкенов можно совместить, если алкен обрабатывать водной 30-70%-ной перекисью водорода в муравьиной или трифторуксусной кислоте. Обе эти кислоты являются достаточно сильными для того, чтобы вызвать раскрытие оксиранового цикла.

Раскрытие оксиранового кольца, катализируемое основанием, также приводит к образованию циклических транс -гликолей.

Следовательно, двухстадийный процесс эпоксидирования алкенов с последующим щелочным гидролизом эпоксидов также является реакцией анти -гидроксилирования алкенов.
3. син -Гидроксилирование
Некоторые соли и оксиды переходных металлов в высших степенях окисления являются эффективными реагентами син -гидроксилирования двойной связи алкена, когда обе гидроксильные группы присоединяются с одной и той же стороны двойной связи. Окисление алкенов перманганатом калия - один из старейших методов син -гидроксилирования двойной связи - продолжает широко использоваться, несмотря на свойственные ему ограничения. Цис -1,2-циклогександиол был впервые получен В.В. Марковниковым в 1878 году гидроксилированием циклогексена водным раствором перманганата калия при 0 0 С.

Этот метод в дальнейшем получил развитие в работах русского ученого Е.Е. Вагнера, поэтому син -гидроксилирование алкенов под действием водного раствора перманганата калия носит название реакции Вагнера. Перманганат калия является сильным окислителем, способным не только гидроксилировать двойную связь, но и расщеплять образующийся вицинальный диол. Для того, чтобы по возможности избежать дальнейшего расщепления гликолей, необходимо тщательно контролировать условия реакции. Выходы гликолей при этом обычно невелики (30-60%). Наилучшие результаты достигаются при гидроксилировании алкенов в слабощелочной среде (рН~8 9) при 0-5 0 С разбавленным 1%-ным водным раствором KMnO 4 .


Первоначально при окислении алкенов перманганатом калия образуется циклический эфир марганцевой кислоты, который немедленно гидролизуется до вицинального диола.

Циклический эфир марганцевой кислоты как интермедиат не был выделен, однако его образование следует из экспериментов с меченым 18 О перманганатом калия: оба атома кислорода в гликоле оказываются мечеными при окислении алкена KMn 18 O 4 . Это означает, что оба атома кислорода переходят от окислителя, а не из растворителя - воды, что находится в хорошем соответствии с предлагаемым механизмом.
Другой метод син -гидроксилирования алкенов под действием оксида осмия (VIII) OsO 4 был предложен Р. Криге в 1936 году. Тетраоксид осмия представляет собой бесцветное, летучее, кристаллическое вещество, хорошо растворимое в эфире, диоксане, пиридине и др. органических растворителях. При взаимодействии тетраоксида осмия с алкенами в эфире или диоксане образуется черный осадок циклического эфира осмиевой кислоты - осмат, который легко может быть изолирован в индивидуальном виде. Присоединение OsO 4 к двойной связи заметно ускоряется в растворе в пиридине. Разложение осматов до вицинальных гликолей достигается действием водного раствора гидросульфита натрия или сероводородом.

Выходы продуктов син -гидроксилирования алкенов в этом методе значительно выше, чем при использовании перманганата в качестве окислителя. Важным достоинством метода Криге является отсутствие продуктов окислительного расщепления алкенов, характерного для перманганатного окисления.


Тетраоксид осмия очень дорогой и труднодоступный реагент, к тому же он токсичен. Поэтому оксид осмия (VIII) используется при синтезе малых количеств трудно доступных веществ с целью получения наиболее высокого выхода диола. С целью упрощения син -гидроксилирования алкенов под действием OsO 4 была разработана методика, позволяющая использовать лишь каталитические количества этого реагента. Гидроксилирование алкенов осуществляется с помощью перекиси водорода в присутствии OsO 4 , например:

В заключение этого раздела приведем стереохимические отношения между алкеном цис - или транс -конфигурации и конфигурацией образующегося вицинального диола, который может быть цис - или транс -изомером, эритро - или трео -формой, мезо - или D,L -формой в зависимости от заместителей в алкене:

Аналогичные стереохимические отношения наблюдаются и в других реакциях син - или анти -присоединения по кратной связи водорода, галогенводородов, воды, галогенов, гидридов бора и др. реагентов.
Данный материал может быть сложен в освоении при самостоятельном обучении, ввиду большого объема информации, многих нюансов, всевозможных НО и ЕСЛИ. Читать внимательно!
О чем именно пойдет речь?
Помимо полного окисления (горения), для некоторых классов органических соединений характерны реакции неполного окисления, при этом они превращаются в другие классы.
Существуют специфические окислители для каждых классов: CuO(для спиртов),Cu(OH) 2 и OH (для альдегидов) и другие.
Но есть два классических окислителя, которые, если так можно выразиться, универсальные для многих классов.
Это перманганат калия – KMnO 4 . И бихромат (дихромат) калия – K 2 Cr 2 O 7 . Эти вещества являются сильными окислителями за счет марганца в степени окисления +7, и хрома в степени окисления +6, соответственно.
Реакции с этими окислителями встречаются довольно часто, однако нигде нет целостного руководства, по какому принципу выбирать продукты таких реакций.
На практике действует очень много факторов, влияющих на ход реакции (температура, среда, концентрация реагентов и т.д.). Часто получается смесь продуктов. Поэтому предугадать продукт, который образуется практически невозможно.
А для ЕГЭ это не годится: там нельзя написать «может быть или так, или вот так, или иначе, или смесь продуктов». Там нужна конкретика.
Составители заданий вложили определенную логику, определенный принцип по которому следует писать определенный продукт. К сожалению, они ни с кем не поделились.
Данный вопрос в большинстве пособий довольно скользко обходится стороной: в качестве примера приведено две-три реакции.
Представляю в этой статье, то, что можно назвать результатами исследования-анализа заданий ЕГЭ. Логика и принципы составления реакций окисления перманганатом и дихроматом разгадана довольно с высокой точностью (в соответствии со стандартами ЕГЭ). Обо всем по порядку.
Определение степени окисления .
Первое, когда имеем дело с окислительно-восстановительными реакциями, всегда есть окислитель и восстановитель.
Окислителем является марганец в перманганате или хром в дихромате, восстановителем – атомы в органике (а именно – атомы углерода).
Мало определить продукты, реакция должна быть уравнена. Для уравнивания традиционно используют метод электронного баланса. Для применения этого метода необходимо определить степени окисления восстановителей и окислителей до и после реакции.
У неорганических веществ степени окисления умеем с 9 класса:
А вот в органике, наверное, в 9 классе не определяли. Поэтому прежде, чем научиться писать ОВР в органической химии, нужно научиться определять степень окисления углерода в органических веществах. Делается это немного по-другому, иначе чем в неорганической химии.
У углерода максимальная степень окисления +4, минимальная -4. И он может проявлять любую степень окисления этого промежутка: -4, -3, -2, -1, 0, +1, +2, +3, +4.
Для начала нужно вспомнить, что такое степень окисления.
Степень окисления – это условный заряд, возникающий на атоме, при допущении, что электронные пары смещаются полностью в сторону более электроотрицательного атома.
Поэтому степень окисления определяется числом смещенных электронных пар: если она смещается к данному атому, то он приобретает избыточный минус(-) заряд, если от атома, то он приобретает избыточный плюс(+) заряд. В принципе это вся теория, которую нужно знать, для определения степени окисления атома углерода.
Для определения степени окисления конкретного атому углерода в соединении нам нужно рассмотреть КАЖДУЮ его связь и посмотреть в какую сторону будет смещаться электронная пара и какой избыточный заряд (+ или -) будет от этого возникать на атоме углерода.
Разберем конкретные примеры:
У углерода три связи с водородом . Углерод и водород – кто более электроотрицателен? Углерод, значит, по этим трем связям электронная пара будет смещаться в сторону углерода. Углерод забирает у каждого водорода по одному отрицательному заряду: получается -3
Четвертая связь с хлором. Углерод и хлор – кто более электроотрицателен? Хлор, значит, по этой связи электронная пара будет смещаться в сторону хлора. У углерода появляется один положительный заряд +1.
Затем, нужно просто сложить: -3 + 1 = -2. Степень окисления этого атома углерода: -2.

Определим степень окисления каждого атома углерода:

У углерода три связи с водородом. Углерод и водород – кто более электроотрицателен? Углерод, значит, по этим трем связям электронная пара будет смещаться в сторону углерода. Углерод забирает у каждого водорода по одному отрицательному заряду: получается -3
И еще одна связь с другим углеродом. Углерод и другой углерод – их электроотрицательности равны, поэтому смещения электронной пары не происходит (связь не полярная).

У этого атома две связи с одним атомом кислорода, и еще одна связь с другим атомом кислорода (в составе группы OH). Более электроотрицательные атомы кислорода по трем связям оттягивают на себя электронную пару у углерода, у углерода появляется заряд +3.
Четвертой связью углерод связан с другим углеродом, как мы уже говорили, по этой связи электронная пара не смещается.


Двумя связями углерод связан с атомами водорода. Углерод, как более электроотрицательный оттягивает себе по одной паре электронов по каждой связи с водородом, приобретает заряд -2.
Двойной связью углерода связан с атомом кислорода. Более электроотрицательный кислород оттягивает на себя по каждой связи одну электронную пару. Вместе получается у углерода оттягивается две электронные пары. Углерод приобретает заряд +2.
Вместе получается +2 -2 = 0.

Определим степень окисления вот этого атома углерода:
Тройная связь с более электроотрицательным азотом – дает углероду заряд +3, по связи с углеродом смещения электронной пары не происходит.

Окисление перманганатом.
Что будет с перманаганатом?
Окислительно-восстановительная реакция с перманганатом может протекать в разных средах (нейтральная, щелочная, кислая). И от среды зависит, как именно будет протекать реакция, и какие при этом образуются продукты.
Поэтому может идти по трем направлениям:

Перманганат, являясь окислителем, восстанавливается. Вот продукты его восстановления:
- Кислая среда .
Среду подкисляют серной кислотой (H 2 SO 4). Марганец восстанавливается до степени окисления +2. И продукты восстановления будут:
KMnO 4 + H 2 SO 4 → MnSO 4 + K 2 SO 4 + H 2 O
- Щелочная среда .
Для создания щелочной среды добавляют довольно концентрированную щелочь (KOH). Марганец восстанавливается до степени окисления +6. Продукты восстановления
KMnO 4 + KOH → K 2 MnO 4 + H 2 O
- Нейтральная среда (и слабощелочная ).
В нейтральной среде кроме перманганата в реакцию так же вступает вода (которую мы пишем в левой части уравнения), марганец будет восстанавливаться до +4 (MnO 2), продукты восстановления будут:
KMnO 4 + H 2 O → MnO 2 + KOH
А в слабощелочной среде (в присутствии раствора KOH невысокой концентрации):
KMnO 4 + KOH → MnO 2 + H 2 O

Что будет с органикой?
Первое, что нужно усвоить – все начинается со спирта! Это начальная стадия окисления. Окислению подвергается тот углерод, к которому присоединена гидроксильная группа.
При окислении атом углерода «приобретает» связь с кислородом. Поэтому, когда записывают схему реакции окисления, над стрелкой пишут [O]:
Первичный спирт окисляется сначала до альдегида, потом до карбоновой кислоты:

Окисление вторичного спирта обрывается на второй стадии. Так как углерод находится посередке, образуется кетон, а не альдегид (атом углерода в кетонной группе уже физически не может образовать связь с гидроксильной группой):

Кетоны , третичные спирты и карбоновые кислоты дальше уже не окисляются:

Процесс окисления ступенчатый – пока есть куда окисляться и есть для этого все условия – реакция идет. Все заканчивается продуктом, который в данных условиях не окисляется: третичный спирт, кетон или кислота.
Стоит отметить стадии окисления метанола. Вначале он окисляется до соответствующего альдегида, затем до соответствующей кислоты:

Особенностью этого продукта (муравьиной кислоты) является то, что углерод в карбоксильной группе связан с водородом, и если приглядеться, то можно заметить, что это ни что иное как альдегидная группа:

А альдегидная группа, как мы выяснили ранее, окисляется дальше до карбоксильной:

Узнали полученное вещество? Его брутто-формула H 2 CO 3 . Это угольная кислота, которая распадается на углекислый газ и воду:
H 2 CO 3 → H 2 O + CO 2
Поэтому метанол, муравьиный альдегид и муравьиная кислота (за счет альдегидной группы) окисляются до углекислого газа.
Мягкое окисление.
Мягкое окисление – это окисление без сильного нагревания в нейтральной или слабощелочной среде (над реакцией пишут 0 ° или 20 °) .
Важно помнить, что спирты в мягких условиях не окисляются. Поэтому если они образуются, то на них окисление и останавливается. Какие вещества будут вступать в реакцию мягкого окисления?
- Содержащие двойную связь C=C (Реакция Вагнера).
При этом π-связь разрывается и на освободившиеся связи «садится» по гидроксильной группе. Получается двухатомный спирт:

Напишем реакцию мягкого окисления этилена (этена). Запишем исходные вещества и предскажем продукты. При этом H 2 O и КOH пока не пишем: они могут оказаться как в правой части уравнения, так и в левой. И сразу определяем степени окисления участвующих в ОВР веществ:

Составим электронный баланс (имеем ввиду, что восстановителя два – два атома углерода, окисляются они по-отдельности):

Расставим коэффициенты:

В конце надо дописать недостающие продукты (H 2 O и KOH). Справа не хватает калия – значит щелочь будет справа. Ставим коэффициент перед ней. Слева не хватает водорода, значит, вода слева. Ставим перед ней коэффициент:

Проделаем то же самое с пропиленом (пропеном):


Часто подсовывают циклоалкен. Пусть он вас не смутит. Это обычный углеводород с двойной связью:

Где бы не была эта двойная связь, окисление будет идти одинаково:

- Содержащие альдегидную группу .
Альдегидная группа более реакционноспособная (легче вступает в реакции), чем спиртовая. Поэтому альдегидная будет окисляться. До кислоты:

Рассмотрим на примере ацетальдегида (этаналя). Запишем реагенты и продукты и расставим степени окисления. Составим баланс и поставим коэффициенты перед восстановителем и окислителем:

В нейтральной среде и слабощелочной ход реакции будет немного разным.
В нейтральной среде, как мы помним при этом в левой части уравнения пишем воду, а в правой части уравнения щелочь (образуется в ходе реакции):

При этом в одной смеси оказываются рядом кислота и щелочь. Происходит нейтрализация.

Они не могут существовать рядом и реагируют, образуется соль:
При этом если мы посмотрим на коэффициенты в уравнении, то поймем, что кислоты 3 моля, а щелочи 2 моля. 2 моля щелочи может нейтрализовать только 2 моля кислоты (образуется 2 моля соли). А один моль кислоты остается. Поэтому конечное уравнение будет таким:

В слабощелочной среде щелочь в избытке – ее добавляют до реакции, поэтому нейтрализуется вся кислота:

Похожая ситуация возникает при окислении метаналя. Он, как мы помним, окисляется до углекислого газа:

Нужно иметь ввиду, что оксид углерода (IV) CO 2 кислотный. И будет реагировать с щелочью. И так как угольная кислота двухосновная, может образовываться как кислая соль, так и средняя. Это зависит от соотношения между щелочью и углекислым газом:
Если щелочь относится к углекислому газу как 2:1 , то будет средняя соль:
Или же щелочи может быть значительно больше (больше, чем в два раза). Если ее больше чем в два раза, то будет оставаться остаток щелочи:
3KOH + CO 2 → K 2 CO 3 + H 2 O + KOH
Такое будет возникать в щелочной среде (где щелочи избыток, так как она добавлена в реакционную смесь до реакции) или в нейтральной среде, когда щелочи образуется много.
Но если щелочь относится к углекислому газу как 1:1 , то будет кислая соль:
KOH + CO 2 → KHCO 3
Если углекислого газа больше, чем нужно, то он остается в избытке:
KOH + 2CO 2 → KHCO 3 + CO 2
Такое будет в нейтральной среде, если щелочи образуется мало.
Запишем исходные вещества, продукты, составим баланс, проставим степени окисления перед окислителем, восстановителем и продуктами, которые из них образуются:

В нейтральной среде справа будет образовываться щелочь (4KOH):

Теперь надо понять, что же будет образовываться при взаимодействии трех молей CO 2 и четырех молей щелочи.
3CO 2 + 4KOH → 3KHCO 3 + KOH
KHCO 3 + KOH → K 2 CO 3 + H 2 O
Поэтому получается вот так:
3CO 2 + 4KOH → 2KHCO 3 + K 2 CO 3 + H 2 O
Поэтому в правой части уравнения пишем два моля гидрокарбоната и один моль карбоната :

А в слабощелочной среде таких заморочек нет: из-за того, что щелочи избыток, будет образовываться средняя соль:

То же самое будет при окислении альдегида щавелевой кислоты:

Как и в предыдущем примере, образуется двухосновная кислота, и по уравнению должно получиться 4 моля щелочи (так как 4 моля перманганата).
В нейтральной среде опять-таки, всей щелочи не хватит на полную нейтрализацию всей кислоты.
Три моля щелочи уходит на образование кислой соли, один моль щелочи остается:
3HOOC–COOH + 4KOH → 3KOOC–COOH + KOH
И этот один моль щелочи уходит на взаимодействие с одним молем кислой соли:
KOOC–COOH + KOH → KOOC–COOK + H 2 O
Получается вот так:
3HOOC–COOH + 4KOH → 2KOOC–COOH + KOOC–COOK + H 2 O
Конечное уравнение:

В слабощелочной среде образуется средняя соль из-за избытка щелочи:

- Содержащие тройную связь C ≡ C .
Помните, что было при мягком окислении соединений с двойной связью? Если не помните, то пролистайте назад – вспомните.
π-связь рвется, на атомы углерода прикрепляется по гидроксильной группе. Здесь тот же принцип. Только стоит помнить, что в тройной связи есть две π-связи. Сначала это происходит по первой π-связи:
Потом по другой π-связи:

Структура, в которой у одного атома углерода две гидроксильные группы, крайне неустойчива. Когда в химии что-то не устойчиво, оно стремится, чтобы что-то «отвалилось». Отваливается вода, вот так:

Получается карбонильная группа.
Рассмотрим примеры:
Этин (ацетилен). Рассмотрим стадии окисления этого вещества:

Отщепление воды:




Как и в предыдущем примере, в одной реакционной смеси кислота и щелочь. Происходит нейтрализация – образуется соль. Как видно по коэффициенту перед перманганатом щелочи будет 8 молей, то есть вполне хватает для нейтрализации кислоты. Конечное уравнение:

Рассмотрим окисление бутина-2:

Отщепление воды:

Здесь кислоты не образуется, поэтому морочиться над нейтрализацией не надо.
Уравнение реакции:

Эти различия (между окислением углерода с краю и посередине цепи) ярко демонстрируются на примере пентина:

Отщепление воды:

Получается вещество интересного строения:

Альдегидная группа продолжает окисляться:
Запишем исходные вещества, продукты, определим степени окисления, составим баланс, проставим коэффициенты перед окислителем и восстановителем:

Щелочи должно образовываться 2 моля (так как коэффициент перед перманганатом 2), следовательно, вся кислота нейтрализуется:

Жесткое окисление .
Жесткое окисление – это окисление в кислой , сильнощелочной среде. А также, в нейтральной (или слабощелочной), но при нагревании .
В кислой среде тоже иногда нагревают. Но чтобы жесткое окисление пошло не в кислой среде, нагревание – обязательное условие.
Какие вещества будут подвергаться жесткому окислению? (Вначале разберем только в кислой среде – а потом дополним нюансами, которые возникают при окислении в сильнощелочной и нейтральной или слабощелочной (при нагревании) среде).
При жестком окислении процесс идет по максимуму. Пока есть, что окисляться – окисление идет.
- Спирты. Альдегиды .
Рассмотрим окисление этанола. Поступенчато он окисляется до кислоты:

Записываем уравнение. Записываем исходные вещества, продукты ОВР, проставляем степени окисления, составляем баланс. Уравниваем реакцию:

Если реакцию проводить при температуре кипения альдегида, когда он будет образовываться, то будет испаряться (улетать) из реакционной смеси, не успевая окисляться дальше. Того же эффекта можно добиться в очень щадящих условиях (слабое нагревание). В этом случае в качестве продукта пишем альдегид:

Рассмотрим окисление вторичного спирта на примере пропанола-2. Как уже было сказано, окисление обрывается на втором этапе (образование карбонильного соединения). Так как образуется кетон, который не окисляется. Уравнение реакции:

Окисление альдегидов рассмотрим на примете этаналя. Он тоже окисляется до кислоты:

Уравнение реакции:

Метаналь и метанол, как было сказано ранее, окисляются до углекислого газа:

Метаналь:

- Содержащие кратные связи .
При этом происходит разрыв цепи по кратной связи. И атомы, которые образовывали ее подвергаются окислению (приобретают связь с кислородом). Окисляются насколько это возможно.
При разрыве двойной связи из обрывков образуются карбонильные соединения (в схеме ниже: из одного обрывка – альдегид, из другого – кетон)

![]()
Разберем окисление пентена-2:

Окисление «обрывков»:

Получается, что образуется две кислоты. Запишем исходные вещества и продукты. Определим степени окисления у атомов, которые ее меняют, составим баланс, уравняем реакцию:
Составляя электронный баланс, имеем ввиду, что восстановителя два – два атома углерода, окисляются они по-отдельности:

Не всегда будет образовываться кислота. Разберем, например, окисление 2-метилбутена:

Уравнение реакции:

Абсолютно тот же самый принцип при окислении соединений с тройной связью (только окисление идет сразу с образованием кислоты, без промежуточного образования альдегида):

Уравнение реакции:

Когда кратная связь расположена ровно посередине, то получается не два продукта, а один. Так как «обрывки» одинаковые и окисляются они до одинаковых продуктов:

Уравнение реакции:

- Дважды коронованная кислота .
Есть одна кислота, у которой карбоксильные группы (короны) соединены друг с другом:
Это щавелевая кислота. Две короны рядом трудно уживаются. Она конечно устойчива в обычных условиях. Но из-за того, что в ней две карбоксильные группы соединены друг с другом, она менее устойчивая, чем другие карбоновые кислоты.
И поэтому при особо жестких условиях она может быть окислена. Происходит разрыв связи между «двумя коронами»:

Уравнение реакции:

- Гомологи бензола (и их производные) .
Сам бензол не окисляется, из-за того, что ароматичность делает эту структуру очень устойчивой
А вот его гомологи окисляются. При этом тоже происходит разрыв цепи, главное знать где именно. Действуют некоторые принципы:
- Бензольное кольцо само не разрушается, и остается целым до конца, разрыв связи происходит в радикале.
- Окисляется атом, непосредственно связанный с бензольным кольцом. Если после него углеродная цепь в радикале продолжается – то разрыв будет после него.


Разберем окисление метилбензола. Там окисляется один атом углерода в радикале:
Уравнение реакции:

Разберем окисление изобутилбензола:

Уравнение реакции:

Разберем окисление втор-бутилбензола:

Уравнение реакции:

При окислении гомологов бензола (и производных гомологов) с несколькими радикалами, образуются двух- трех- и более основные ароматические кислоты. Например, окисление 1,2-диметилбензола:

Производные гомологов бензола (в которых у бензольного кольца есть не углеводородные радикалы), окисляются так же. Другая функциональная группа у бензольного кольца не мешает:


Промежуточный итог. Алгоритм «как записать реакцию жесткого окисления перманганатом в кислой среде»:
- Записать исходные вещества (органика + KMnO 4 + H 2 SO 4).
- Записать продукты окисления органики (окисляться будут соединения содержащие спиртовую, альдегидную группы, кратные связи, а также гомологи бензола).
- Записать продукт восстановления перманганата (MnSO 4 + K 2 SO 4 + H 2 O).
- Определить степени окисления у участников ОВР. Составить баланс. Проставить коэффициенты у окислителя и восстановителя, а также у веществ, которые из них образуются.
- Затем рекомендовано посчитать сколько сульфат-анионов в правой части уравнения, в соответствии с этим поставить коэффициент перед серной кислотой слева.
- В конце поставить коэффициент перед водой.
Жесткое окисление в сильнощелочной среде и нейтральной или слабощелочной (при нагревании) среде .
Эти реакции встречаются гораздо реже. Можно сказать, что такие реакции – это экзотика. И как положено любым экзотическим реакциям, эти оказались самыми противоречивыми.
Жесткое окисление оно и в Африке жесткое, поэтому органика окисляется так же, как и в кислой среде.
Отдельно реакции для каждого класса разбирать не будем, так как общий принцип уже изложен ранее. Разберем только нюансы.
Сильнощелочная среда :
В сильнощелочной среде перманганат восстанавливается до степени окисления +6 (манганат калия):
KMnO 4 + KOH → K 2 MnO 4 .
В сильнощелочной среде щелочи всегда избыток, поэтому будет проходить полная нейтрализация: если образуется углекислый газ – будет карбонат, если образуется кислота – будет соль (если кислота многоосновная – средняя соль).
Например, окисление пропена:

Окисление этилбензола:

Слабощелочная или нейтральная среда при нагревании :
Здесь также необходимо всегда учитывать возможность нейтрализации.
Если окисление протекает в нейтральной среде и образуется кислотное соединение (кислота или углекислый газ), то образующаяся щелочь будет нейтрализовать это кислотное соединение. Но не всегда щелочи хватит на полную нейтрализацию кислоты.
При окислении альдегидов, например, ее не хватает (окисление будет протекать так же, как и в мягких условиях – температура просто ускорит реакцию). Поэтому образуется и соль, и кислота (оставшаяся грубо говоря в избытке).
Мы это обсуждали, когда разбирали мягкое окисление альдегидов.
Поэтому если у вас образуется кислота в нейтральной среде, нужно внимательно посмотреть хватит ли ее на нейтрализацию всей кислоты. Особое внимание нужно уделить нейтрализации многоосновных кислот.
В слабощелочной среде из-за достаточного количества щелочи образуются только средние соли, так, как щелочи избыток.
Как правило, щелочи при окислении в нейтральной среде вполне хватает. И уравнение реакции что в нейтральной, что в слабощелочной среде будут одинаковы.
Для примера разберем окисление этилбензола:


Щелочи вполне хватает на полную нейтрализацию полученных кислотных соединений, даже лишнего останется:

Расходуется 3 моля щелочи – 1 остается.
Конечное уравнение:

Эта реакция в нейтральной и слабощелочной среде будет идти одинаково (в слабощелочной среде слева щелочи нет, но это не значит, что ее нет, просто она в реакцию не вступает).
Окислительно-восстановительные реакции с участием дихромата (бихромата) калия.
Бихромат не имеет такого большого разнообразия реакций окисления органики в ЕГЭ.
Окисление бихроматом проводится как правило только в кислой среде. При это хром восстанавливается до +3. Продукты восстановления:
Окисление будет жестким. Реакция будет очень похожа на окисление перманганатом. Окисляться будут те же вещества, что окисляются перманганатом в кислой среде, образовываться будут те же продукты.
Разберем некоторые реакции.
Рассмотрим окисление спирта. Если проводить окисление при температуре кипения альдегида, то он будет уходить их реакционной смеси, не подвергаясь окислению:

В противном случае, спирт может быть напрямую окислен до кислоты.
Альдегид, полученный в ходе предыдущей реакции, можно «поймать», и заставить его окисляться до кислоты:

Окисление циклогексанола. Циклогексанол является вторичным спиртом, поэтому образуется кетон:

Если тяжело определить степени окисления атомов углерода по такой формуле, на черновике можно расписать:

Уравнение реакции:

Рассмотрим окисление циклопентена.
Двойная связь рвется (цикл размыкается), атомы, которые ее образовывали окисляются до максимума (в данном случае, до карбоксильной группы):


Некоторые особенности окисления в ЕГЭ, с которыми мы не совсем согласны.
Те «правила», принципы и реакции, которые будут рассмотрены в этом разделе, мы считаем не совсем корректными. Они противоречат не только реальному положению дел (химии как науке), но и внутренней логике школьной программы и ЕГЭ в частности.
Но тем не менее, мы вынуждены дать этот материал именно в том виде, который требует ЕГЭ.
Речь пойдет именно о ЖЕСТКОМ окислении.
Помните, как окисляются гомологи бензола и их произсодные в жестких условиях? Радикалы все обрываются – образуются карбоксильные группы. Обрывки подвергаются окислению уже «самостоятельно»:

Так вот, если вдруг радикале появляется гидроксильная группа, или кратная связь, нужно забыть, что там есть бензольное кольцо. Реакция пойдет ТОЛЬКО по этой функциональной группе (или кратной связи).
Функциональная группа и кратная связь главнее бензольного кольца.

Разберем окисление каждого вещества:
Первое вещество:

Нужно не обращать внимание на то, что есть бензольное кольцо. С точки зрения ЕГЭ – это всего лишь вторичный спирт. Вторичные спирты окисляются до кетонов, а кетоны далее не окисляются:

Пусть это вещество у нас будет окисляться бихроматом:

Второе вещество:

Это вещество окисляется, просто как соединение с двойной связью (на бензольное кольцо не обращаем внимание):

Пусть оно будет окисляться в нейтральном перманганате при нагревании:

Образовавшейся щелочи хватает на полную нейтрализацию углекислого газа:
2KOH + CO 2 → K 2 CO 3 + H 2 O
Итоговое уравнение:

Окисление третьего вещества:

Пусть окисление будет протекать перманганатом калия в кислой среде:

Окисление четвертого вещества:

Оно пусть окисляется в сильнощелочной среде. Уравнение реакции будет:

Ну и напоследок, вот так окисляется винилбензол:

А окисляется он до бензойной кислоты, нужно иметь ввиду, что по логике ЕГЭ он так окисляется не потому, что он – производное бензола. А потому, что он содержит двойную связь.
Заключение .
Это все, что нужно знать об окислительно-восстановительных реакциях с участием перманганата и бихромата в органике.
Не удивляйтесь если, некоторые моменты изложенные в данной статье, вы слышите впервые. Как уже было сказано, тема эта очень обширная и противоречивая. И несмотря на это почему-то ей уделяется крайне мало внимания.
Как вы, возможно, убедились, двумя-тремя реакциями не объяснить всех закономерностей этих реакций. Здесь нужен комплексный подход и подробное объяснения всех моментов. К сожалению в учебниках и на интернет ресурсах тема раскрыта не полностью, либо не раскрыта совсем.
Я постарался устранить эти недоработки и недочеты и рассмотреть эту тему целиком, а не частично. Надеюсь, мне это удалось.
Благодарю Вас за внимание, всего Вам хорошего! Успехов в освоении химической науки и сдаче экзаменов!
Санкт-Петербургский Государственный Технологический Институт
(Технический Университет)
Кафедра органической химии Факультет 4
Группа 476
Курсовая работа
Окисление алкенов
Студентка………………………………………Рытина А.И.
Преподаватель………………………………... Питерская Ю.Л.
Санкт-Петербург
Введение
1.Эпоксидирование (реакция Н.А. Прилежаева,1909 г.)
2.Гидроксилирование
2.1анти -Гидроксилирование
2.2син -Гидроксилирование
3.Окислительное расщепление алкенов
4.Озонолиз
5.Окисление алкенов в присутствии солей палладия
Заключение
Список использованных источников
Введение
Окисление - одно из наиболее важных и распространенных превращений органических соединений.
Под окислением в органической химии понимают процессы, приводящие к обеднению соединения водородом или обогащению его кислородом. При этом происходит отнятие от молекулы электронов. Соответственно, под восстановлением понимают отрыв от органической молекулы кислорода или присоединение к ней водорода.
В окислительно-восстановительных реакциях окислителями являются соединения, обладающие большим сродством к электрону (электрофилы), а восстановителями – соединения, имеющие склонность к отдаче электронов (нуклеофилы). Легкость окисления соединения возрастает вместе с ростом его нуклеофильности.
При окислении органических соединений, как правило, полной передачи электронов и соответственно изменения валентности атомов углерода не происходит. Поэтому понятие степени окисления – условного заряда атома в молекуле, вычисленного, исходя из предположения, что молекула состоит только из ионов – носит лишь условный, формальный характер.
При составлении уравнений окислительно-восстановительных реакций необходимо определить восстановитель, окислитель и число отдаваемых и принимаемых электронов. Как правило, коэффициенты подбирают, используя метод электронно-ионного баланса (метод полуреакций).
В этом методе рассматривают переход электронов от одних атомов или ионов к другим с учетом характера среды (кислая, щелочная или нейтральная), в которой протекает реакция. Для уравнивания числа атомов кислорода и водорода вводят или молекулы воды и протоны (если среда кислая), или молекулы воды и гидроксид-ионы (если среда щелочная).
Таким образом, при написании полуреакций восстановления и окисления нужно исходить из состава ионов, действительно имеющихся в растворе. Вещества малодиссоциирующие, плохо растворимые или выделяющиеся в виде газа следует писать в молекулярной форме.
В качестве примера рассмотрим процесс окисления этилена разбавленным водным раствором перманганата калия (реакция Вагнера). В ходе данной реакции этилен окисляется до этиленгликоля, а перманганат калия восстанавливается до диоксида марганца. По месту двойной связи присоединяются два гидроксила :
3С 2 H 4 + 2KMnO 4 +4H 2 O→ 3C 2 H 6 O 2 + 2MnO 2 +2KOH
Полуреакция восстановления: MnO 4 ¯ + 2H 2 O + 3 e → MnO 2 + 4OH ¯ 2Полуреакция окисления: С 2 H 4 + 2OH − − 2 e → C 2 H 6 O 2 3
Окончательно имеем в ионном виде:
2MnO 4 ¯ + 4H 2 O + 3C 2 H 4 + 6OH ¯ → 2MnO 2 + 8OH ¯ + 3C 2 H 6 O 2
После проведения необходимых сокращений подобных членов, записываем уравнение в молекулярном виде:
3C 2 H 4 + 2KMnO 4 + 4 H 2 O = 3C 2 H 6 O 2 + 2MnO 2 + 2KOH.
Характеристика некоторых окислителей
Кислород
Кислород воздуха находит широкое применение в технологических процессах, так как является наиболее дешевым окислителем. Но окисление кислородом воздуха сопряжено с трудностями, связанными с контролем процесса, который протекает в различных направлениях. Окисление обычно проводят при высокой температуре в присутствии катализаторов.
Озон
Озон O 3 применяют для получения альдегидов и кетонов, если их затруднительно получить другими способами. Чаще всего озон применяют для установления структуры ненасыщенных соединений. Получают озон при действии тихого электрического разряда на кислород. Одним из существенных достоинств озонирования, по сравнению с хлорированием, является отсутствие токсинов после обработки .
Перманганат калия
Перманганат калия – наиболее часто применяемый окислитель. Реактив растворим в воде (6.0% при 20ºС), а также в метаноле, ацетоне и уксусной кислоте. Для окисления применяют водные (иногда ацетоновые) растворы KMnO 4 в нейтральной, кислой или щелочной среде. При проведении процесса в нейтральной среде в реакционную массу добавляют соли магния, алюминия или пропускают углекислый газ для нейтрализации выделяющегося во время реакции гидроксида калия. Реакцию окисления KMnO 4 в кислой среде чаще всего ведут в присутствии серной кислоты. Щелочную среду при окислении создает образующийся во время реакции KOH, либо его изначально добавляют в реакционную массу. В слабощелочной и нейтральной средах KMnO 4 окисляет по уравнению:
KMnO 4 + 3 e + 2H 2 O = K + + MnO 2 + 4OH ¯
в кислой среде:
KMnO 4 + 5 e + 8H + = K + + Mn 2+ + 4H 2 O
Перманганат калия используется для получения 1,2-диолов из алкенов, при окислении первичных спиртов, альдегидов и алкиларенов до карбоновых кислот, а также для окислительного расщепления углеродного скелета по кратным связям.
На практике обычно используется довольно большой избыток (более чем 100%) KMnO 4 . Это объясняется тем, что в обычных условиях KMnO 4 частично разлагается на диоксид марганца с выделением O 2 . Разлагается концентрированной H 2 SO 4 при нагревании в присутствии восстановителей со взрывом; смеси калия перманганата с органическими веществами также взрывчаты .
Надкислоты
Перуксусную и пермуравьиную кислоты получают реакцией 25-90%-ного пероксида водорода с соответствующей карбоновой кислотой по следующей реакции:
RCOOH + H 2 O 2 = RCOOOH + H 2 O
В случае уксусной кислоты это равновесие устанавливается относительно медленно, и для ускорения образования перкислоты обычно в качестве катализатора добавляют серную кислоту. Муравьиная кислота достаточно сильна сама по себе для того, чтобы обеспечить быстрое установление равновесия.
Пертрифторуксусная кислота, получаемая в смеси с трифторуксусной кислотой реакцией трифторуксусного ангидрида с 90%-ным пероксидом водорода, еще более сильный окислитель. Аналогичным образом из уксусного ангидрида и пероксида водорода можно получить перуксусную кислоту.
Особой популярностью пользуется твердая м -хлорпербензойная кислота, поскольку она относительно безопасна в обращении, достаточно стабильна и может храниться длительное время.
Окисление происходит за счет выделяющегося атома кислорода:
RCOOOH = RCOOH + [O]
Надкислоты применяют для получения эпоксидов из алкенов, а также лактонов из алициклических кетонов.
Пероксид водорода
Пероксид водорода – бесцветная жидкость,cмешивается с водой, этанолом и диэтиловым эфиром. 30%-ный раствор H 2 O 2 называется пергидролем. Высококонцентрированный препарат может реагировать с органическими веществами со взрывом. При хранении разлагается на кислород и воду. Стойкость пероксида водорода возрастает с разбавлением. Для окисления применяют водные растворы различной концентрации (от 3 до 90%) в нейтральной, кислой или щелочной средах.
H 2 O 2 = H 2 O + [O]
Действием этого реагента на α,β-непредельные карбонильные соединения в щелочной среде получают соответствующие эпоксиальдегиды и кетоны, окислением карбоновых кислот в кислой среде синтезируют надкислоты. 30%-ный раствор H 2 O 2 в уксусной кислоте окисляет алкены в 1,2-диолы. Пероксид водорода применяют: для получения органических и неорганических пероксидов, пербората и перкарбоната Na; как окислитель в ракетных топливах; при получении эпоксидов, гидрохинона, пирокатехина, этиленгликоля, глицерина, ускорителей вулканизации группы тиурама и др.; для отбеливания масел, жиров, меха, кожи, текстильных материалов, бумаги; для очистки германиевых и кремниевых полупроводниковых материалов; как дезинфицирующее средство для обезвреживания бытовых и индустриальных сточных вод; в медицине; как источник О 2 в подводных лодках; Н 2 О 2 входит в состав реактива Фентона (Fe 2 + + Н 2 О 2), который используют как источник свободных радикалов ОН в органическом синтезе .
Тетраоксиды рутения и осмия
Тетраоксид осмия OsO 4 – порошок от белого до бледно-желтого цвета с т. пл. 40.6ºС; т. кип. 131.2ºС. Возгоняется уже при комнатной температуре, растворим в воде (7.47 г в 100 мл при 25ºС), ССl 4 (250 г в 100 г растворителя при 20ºС). В присутствии органических соединений чернеет вследствие восстановления до OsO 2 .
RuO 4 представляет собой золотисто-желтые призмы с т. пл. 25.4ºС, заметно возгоняется при комнатной температуре. Умеренно растворим в воде (2.03 г в 100 мл при 20ºС), очень хорошо растворим в CCl 4 . Более сильный окислитель, чем OsO 4 . Выше 100ºС взрывается. Как и тетраоксид осмия обладает большой токсичностью и высокой стоимостью.
Данные окислители применяются для окисления алкенов в α-гликоли в мягких условиях.